







The cost of therapy was recognized as the greatest potential barrier to using gene therapy treatments



It’s important for children to be tested in an appropriate environment


A genetic diagnosis is needed if accurate genetic counselling is desired


If we can be accurate with our phenotyping, we can refer patients for appropriate testing and possibly a definitive diagnosis

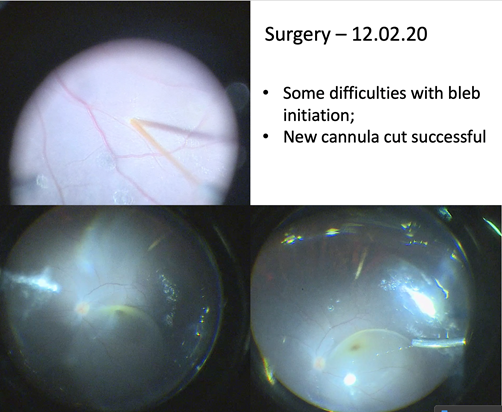
(Courtesy of Robert Henderson MBBS, MD, FRCOphth)

(Courtesy of Robert Henderson MBBS, MD, FRCOphth)
New studies are in progress utilizing antisense oligonucleotides or gene editing technologies such as CRISPR

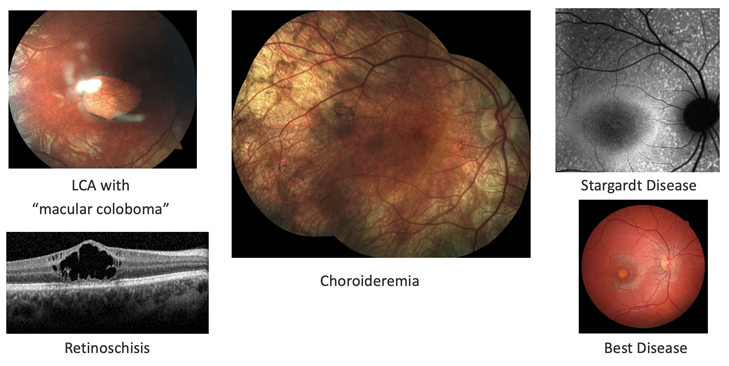
(Courtesy of Hannah Scanga, MS, CGC)
Psychological impacts can be quite vast and affect children and their parents very differently


