
World Society of Pediatric Ophthalmology and Strabismus
Emerging Gene Therapy Options for
the Treatment of Inherited Retinal
Disease in the Paediatric Patient

Table of Contents

- Assessing Baseline Trends in The Treatment of Inherited Retinal Diseases of Paediatric Patients - Ken Nischal, MD
- Diagnosis of Inherited Retinal Diseases – Electrophysiology and Retinal Imaging - Alki Liasis, BSc, PhD
- The Availability of Genetic Testing - Arif O. Khan, MD
- The Role of the Ophthalmologist in Early Detection - David Mackey, MD
- Current and Evolving Paediatric Gene Therapy Options - Robert Henderson MBBS, MD, FRCOphth
- The Essential Role of Counselling - Hannah Scanga, MS, CGC
- Thinking of The Child as a Whole - Eduardo D. Silva, MD, PhD, FEBO

Assessing Baseline Trends in The Treatment of Inherited
Retinal Diseases of Paediatric Patients


Ken Nischal, MD
Department of Ophthalmology
University of Pittsburgh School of Medicine
nischalkk@upmc.edu
Two recent surveys give an indication of the current trends in treating inherited retinal diseases of paediatric patients.
EURETINA Clinical Trends Survey
EURETINA performed a clinical trends survey in Fall 2019, both at the EURETINA Congress in Paris and online. There were 128 questions, and 1,043 EURETINA delegates answered. This is the third year this survey has been conducted1.
The survey found that 74% of delegates have only a moderate to little understanding of gene therapy (Fig.1). And yet, 63% of the same delegates believe that gene therapies are the future for inherited and acquired retinal disorders (Fig.2).
Click to expand
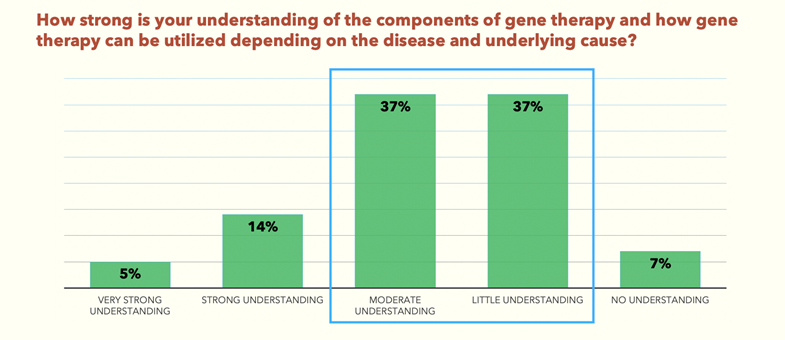
Click to expand
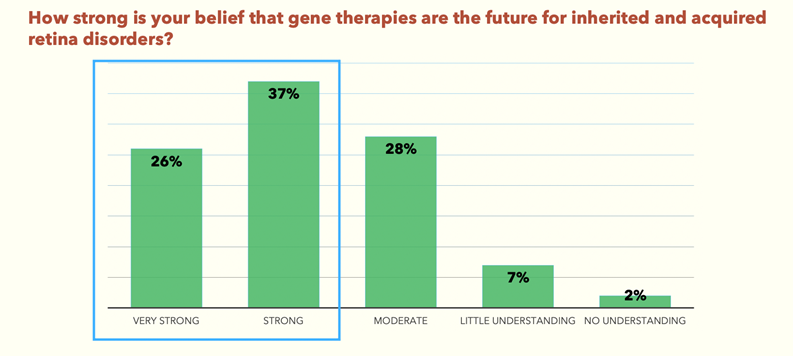
Gene Therapy Survey
Another survey was conducted by Frontline Medical Communications, in collaboration with the National Organization for Rare Disorders (NORD), to learn about knowledge gaps and educational opportunities in gene therapy2. This was a 26-question survey of healthcare providers in 14 specialties, throughout the United States. It was fielded between November 14, 2019 and January 23, 2020, and 1,472 responses were received, for a 95% confidence level.
Sixty-three percent of the NORD survey respondents said they were unaware of current FDA-approved gene therapies, including Luxturna (Spark Therapeutics/Novartis Pharmaceuticals), an adeno-associated virus type 2 (AAV2)-based treatment used in treating patients with an inherited form of retinal diseases.

The cost of therapy was recognized as the greatest potential barrier to using gene therapy treatments
Of the four available or developing methods of gene therapy, gene replacement was most widely recognised at 77%, followed by gene silencing (69%), gene editing (60%) and gene addition (51%).
Cost was recognised as the greatest potential barrier to using gene therapy treatments, but respondents said considerations for staff training and the potential burden for patients are also important.
When asked what educational approaches were used to relay information about gene and cell therapies, continued medical education ranked the highest at 74%, followed by specific disease symposia at medical conferences (72%), then expert speaker programmes (71%), and finally medical journals, either print or online (60%).
Data from recent surveys reveal that, while there is an understanding that gene therapy is a good path forward, there is a lack of overall knowledge and doctors need access to more quality education on current and emerging gene therapy technologies.
- 2019 EURETINA Clinical Trends Survey Supplement
- Gene Therapy Survey Highlights: Knowledge Gaps And Educational Opportunities 03/2020 Neurology Reviews’ 6thannual Rare Neurological Disease Special Report
Honoraria and Advisory Role – Carl Zeiss Meditec;
Advisory Role – Nevakar
Assessing Baseline Trends in The Treatment of Inherited
Retinal Diseases of Paediatric Patients

Alki Liasis, BSc, PhD
Clinical Professor of Ophthalmology
Director of the Electrophysiology Service
University of Pittsburgh School of Medicine
liasisa@chp.edu
It’s important for children to be tested in an appropriate environment. We have posters on our walls that children like, and coloured cabinets that don’t look too clinical and we hide as much of our equipment as we can. We meet children in the waiting room and explain the investigations in appropriate language. We ask if they have fears or phobias and work through that.
Pattern ERGs
For pattern electroretinograms (ERGs), a corneal electrode must be used because of the size of the responses (on the order of 3-5uV), which limits the age of the child. There are two components to the response. The P50 comes from the photoreceptors within the macula, and the N95 comes predominantly from the ganglion cells. Using this we can determine if the child has a macula problem or more of an optic nerve problem.
Retinal Function
Using full field flash ERGs enables us to look at general retinal function. We can determine whether responses are normal or abnormal, and if abnormal, whether it’s affecting the rods, cones, or both. Regardless of whether the flash ERG is normal or not, the pattern ERG can be used to see if there’s macular involvement. By modifying the adult protocol using skin electrodes and a handheld strobe, these ERG recordings can be achieved in awake, young infants.
Flash ERG Recordings
For flash ERGs, an adult protocol should be used for any child old enough and able. This involves pupil dilation, corneal electrodes, standardised stimuli using a Ganzfeld stimulator, dark adaptation for 20 minutes before scotopic recordings, then light adaptation for 10 minutes before photopic recordings. This can typically be achieved in children over 6 years old; however, some children can do this at a younger age, and some older ones cannot.
Different types of corneal electrodes can be used, including gold foil, gold lens, DTL, Burian-Allen and ERG-Jet. Laboratories around the world specialise and have expertise in different electrodes. Guidelines from the International Society for Clinical Electrophysiology of Vision provide new laboratories guidelines to stimulus selection and recording parameters.
Some children can cope with the procedure, but usually not the corneal electrode. Research shows that ERGs recorded using skin electrodes are very similar to those using corneal electrodes; it’s purely a scaling factor. ERGs recorded using the skin electrode are much smaller than those recorded using corneal electrodes with a poorer signal-to-noise ratio. Normative data is needed for both skin and corneal electrode data.
Handheld Stimulators
For children unable to cope with a corneal electrode or a Ganzfeld stimulator, handheld stimulators can be used in conjunction with skin electrodes. We have the child look down toward the stimulator, so the eye is in the optimal position to record the ERG responses (Fig.3).
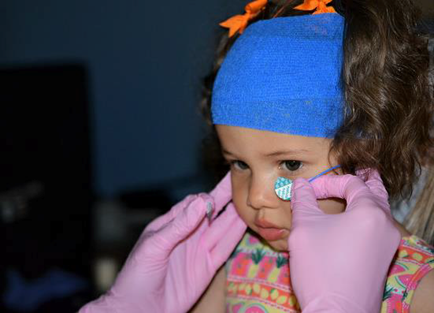

Figure 3: Left panel shows positioning of the self-adhesive electrode, just below the lower eyelid.
Right panel shows the positioning of the child
(Courtesy of Alki Liasis, BSc, PhD )
Coloured filters
In very young patients, we don’t dark adapt, we use coloured filters to preferentially stimulate the rods. For our rod-driven response, we use a dim blue light and then use a standard flash to evoke a mixed-rod ERG, before recording the photopic ERGs. The morphology changes between scotopic and photopic ERGs in adult recordings are evident in the handheld protocol.

It’s important for children to be tested in an appropriate environment
Optical coherence tomography (OCT) images have a near one-to-one relationship with the anatomical features of the retina. Even in a child who is difficult to test, an image slice through the macula typically can be obtained. This provides a lot of information regarding the macula profile and provides information if there are any central areas of macula dysfunction. Quantitative measures can also be extracted. Baseline readings can be determined to be normal or abnormal, and repeat imaging can determine if changes progress over time.
With a cooperative child, a number of scans can be done, allowing segmentation of the retinal layers. This allows us to determine the topography of the various thicknesses of the retina.
Sometimes, if a child is quite difficult to test and we use a low-resolution raster, we’re not able to see the pathology, although some changes may be seen on the fundus image. In a more cooperative child we’re able to do a high-density array of scans to pick up these very small focal changes within the macula.
We also like to do a radial scan around the optic nerve looking at the nerve fiber layer. In some cases, OCT isn’t the best modality for imaging. If an image is not segmented and the nerve fiber layer can’t be picked up, the image really doesn’t provide any information. However, as discussed above, the pattern ERG is able to determine if the photoreceptors within the macula are functioning normally, so for example if an eye has a well-defined P50 and the N95 is degraded and nearly absent, we can attribute the reduced vision to some form of optic nerve pathology.
Conclusion
OCT and electrophysiology both provide valuable information for phenotyping. The OCT provides high spatial definition, in comparison to the electrophysiology, which produces high-detailed temporal resolution able to identify retinal dysfunction. In combination, the two work together in deep phenotyping children with IRDs.
The Availability of Genetic Testing

Arif O. Khan, MD
Section Head of Pediatric Ophthalmology
Director of the Center for Genetic Eye Disease
Professor of Ophthalmology
Eye Institute of Cleveland Clinic, Abu Dhabi
arif.khan@mssm.edu
1. Genetic testing confirms a clinical impression
It is important to first have a clinical impression to direct genetic testing for a patient with an IRD. Molecular diagnosis in children can rule in or rule out risk for associated extraocular disease.
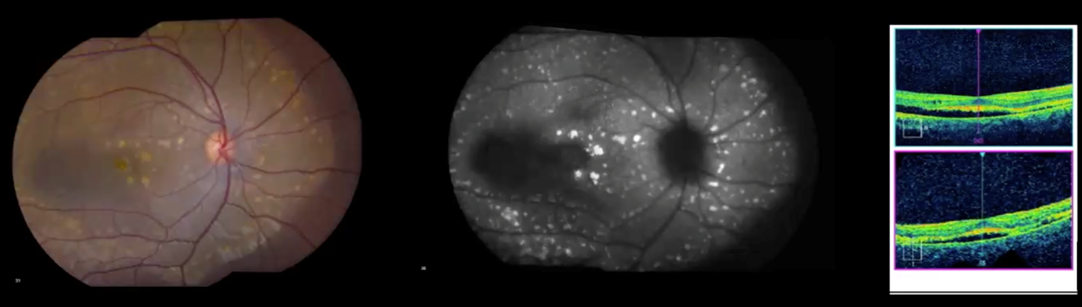
For example, a girl with bilateral findings as shown in Figure 4 was suspected of having an underlying systemic disease and was referred for further evaluation. However, the phenotype of deep subretinal yellow/orange deposits in this distribution associated with subretinal fluid and relatively good vision is almost pathognomonic for autosomal recessive bestrophinopathy. Directed genetic testing of BEST1 confirmed the diagnosis and avoided the suggested systemic work-up because there are no systemic findings associated with the genetic diagnosis.
HAL spectacles can be dispensed in a single visit and there is flexibility in frame selection due to lenslet spacing. A drawback is that they are more costly and are less suitable for co-treatments with higher doses of atropine since there is no reading add. HAL can be impactful in older teens or kids in sports due to visual quality issues.
2. Genetic testing enables genetic counselling
A genetic diagnosis is needed if accurate genetic counselling is desired. One family had two brothers with congenital retinal non-attachment. We diagnosed them with Norrie disease and proved this with genetic testing. The mother was confirmed to be a carrier for this X-linked disease. The mother’s multiple sisters wanted to know if they were carriers for the disease. Genetic testing allowed us to assess carrier status and provide relevant genetic counselling.
A genetic diagnosis is needed if accurate genetic counselling is desired
3. Genetic testing identifies eligibility or non-eligibility for potential treatment
We are in the era of gene therapy and gene therapy trials. Genetic testing is needed to identify eligible patients. One two-year-old boy had nystagmus, sunken globes and poor night vision. He liked to stare at lights. His fundus looked normal, and his ERG was non-recordable. Genetic testing confirmed what his signs and symptoms suggested he possibly had, that is, RPE65-related retinal disease. Once this diagnosis was confirmed he went on to have gene therapy.
4. Genetic testing should be interpreted only by experienced providers
There are nuances to genetic testing that can make it tricky. One girl had been diagnosed with two different retinal diseases at the same time because a next-generation sequencing (NGS) panel predicted that two different retinal genes had biallelic recessive disease-causing variants (CRB1 and AIPL1). But her phenotype, a bullseye maculopathy, was not consistent with biallelic mutations in these two genes. When we did electroretinography (ERG), the signature was pathognomonic for mutations in a specific cone-rod dystrophy gene, KCNV2. But KCNV2 was not on the NGS panel that had been used. We then tested for mutations in KCNV2 separately and a very well-established homozygous mutation was uncovered. Regarding the two biallelic variants in CRB1 and AIPL1, although they were predicted as pathogenic by the program that had been used for the NGS panel, they in fact were not disease causing. Rather, they were just variants. KCNV2 was the causative gene.
5. Genetic testing is challenging in some areas of the world
Barriers can be clinician-related, patient-related or organisation-related. Clinicians are not always familiar with rare genetic disease. Sometimes they have the perception that nothing can be done, but this is no longer the case. Patients may not be aware of their family history, making it harder to realise a genetic diagnosis runs in a family. There is sometimes perceived stigma or guilt of having a genetic diagnosis, and because of this, patients may not want the possibility of gene mutation to be investigated. Some regions lack a referral pathway to providers who are experienced with genetic diseases, and some insurance companies or governments will not cover genetic testing. To combat these challenges, we need to educate clinicians, patients and organisations.
Consultant – Novartis
The Role of the Ophthalmologist in Early Detection

David Mackey, MD
Professor of Ophthalmology
Lions Eye Institute
University of Western Australia
DavidMackey@lei.org.au
The ophthalmologist’s role involves caring for the whole patient and the whole family; coordinating care through geneticists, paediatricians and other specialists, with rehabilitation and education services; and most crucially, managing expectations, particularly when a parent may ask: “Why can’t my child have gene therapy?”
As awareness of genetic testing has increased, patients and families with inherited retinal diseases (IRDs) have begun requesting ophthalmologists refer them for genetic testing. Ophthalmologists need to focus on ordering the most appropriate testing for the most appropriate patients, while keeping in mind how the results would potentially (or not) change management of the patient.
Genetic Testing
Ophthalmologists have been phenotyping IRDs since the 1850s, and many syndromes are clearly characterised by a thorough ophthalmological assessment. Beginning in the 1980s there were a couple decades of exciting gene discovery; more than 250 genes have been identified and listed on the RetNet database.
More and more eye disease-causing genes are going to be discovered worldwide. Papers by Daiger 1 and Huang 2 identified many IRD genes in the pedigrees they studied in the USA and China. A recent paper from the UK 3 has identified that 20 of the most frequent genes account for 72% of families with IRD, and there is considerable overlap of these most common genes with the genetic studies from the US and China. Genetic testing will identify the genes most frequently implicated in large pedigrees, and most common in European or East Asian populations. Therefore, it may be challenging to diagnose individuals from ethnic minorities. However, if we can be accurate with our phenotyping, we can refer patients for appropriate testing and possibly a definitive diagnosis.
There is already gene therapy licensed for RPE65, and many studies are under way. Eligible patients will want to have these treatments and, if possible, be enrolled in genetic research studies.
Clinical Trial Education
The public has been given a lot of information and will demand that ophthalmologists provide treatment, whether pharmacological, gene therapy, or cell-based therapies. However, many of these options are still in the experimental phases, so patients will require explanations of what appropriately approved and funded treatments are available within the setting of registered, ethical, IRB-approved clinical trials, and will need to be guided through scientifically feasible treatments.
Patients need to learn how an appropriate trial is run so they can recognise something approved for an IRD, and whether they are eligible, what the cost is and how long they may need to be in a host country. A trusted support organisation that is up to date with the latest research and translation can lead them through this.
Who pays?
Even though studies may show a particular treatment works and the clinical evidence is strong, healthy economic support is needed.

If we can be accurate with our phenotyping, we can refer patients for appropriate testing and possibly a definitive diagnosis
Insurers and health systems have to be willing to pay, and accredited genetic testing laboratories need to be available. Patients and their families are effective lobbyists and a revolution in the availability of diagnostic genetic testing is anticipated.
Ultimately, however, who pays? Patients and families? Should insurers and governments pay for these expensive treatments, or do patients need to rely on philanthropy such as crowdfunding?
Prevention Over Treatment
There are other often-overlooked options in genetic diseases beyond treatment. Prevention is better than cure. Predictive DNA testing allows counselling, antenatal testing and pre-implantation genetic diagnosis for in vitro fertilisation.
Clinicians need to be aware that patients with IRDs may be choosing to plan a family. Patients must be informed that testing is available and appropriate, and that it is best done early before conception.
Families may be interested in undergoing antenatal testing, but if they are opposed to terminating a pregnancy, they can consider alternatives such as choosing to adopt. Families can also consider pre-implant genetic diagnosis, but this is very rarely funded.
Rehabilitation
The forgotten revolution in IRDs and associated visual impairment is rehabilitation. Assistive support has come a long way from a dog, a white cane and Braille. We now have technologies that can deal with the major problems of reading, recognising faces, navigating in urban spaces, and driving. Artificial intelligence incorporated in driverless cars and smartphones will be able to compensate for visual impairment.
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013 Aug;84(2):132-41.
- Huang X-F, Huang F, Wu K-C, et al. Genotype-phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med. 2015 Apr;17(4):271-8.
- Pontikos N, Arno G, Jurkute N, et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology. 2020 Oct;127(10):1384-1394.
Current and Evolving Paediatric Gene Therapy Options

Robert Henderson MBBS, MD, FRCOphth
Paediatric Ophthalmic Surgeon
Moorfields Eye Hospital
Great Ormond Street Hospital
London, UK
robert.henderson@gosh.nhs.uk
Gene therapy is an exciting field, but much remains to be learned about it, including different modes of administration, different vectors and reactions, including the immune responses to gene therapy.
Retinal gene replacement therapy aims at reversing an inadequate supply of healthy gene product, in autosomal recessive or X-linked disease states, to rectify cellular morphology and restore normal cell functionality.
There are several trials and one licensed treatment (Luxturna, Spark Therapeutics/Novartis Pharmaceuticals) for a number of retinal diseases. New studies are in progress utilising antisense oligonucleotides or gene editing technologies such as CRISPR.
Trial Participants
Determining the eligibility of patients for trials of these treatments requires detailed phenotyping. Using OCT, electrophysiology and a wide array of visual function tests including age-appropriate acuity, contrast sensitivity, colour testing and visual fields, we can ensure there are viable target cells. But no patient can be enrolled until there is genetic confirmation of disease-associated alleles together with an observed phenotype that fits the genetic condition.
Voretigene neparvovec (Luxturna, Spark Therapeutics/Novartis Pharmaceuticals) is a licensed gene therapy replacement for patients with biallelic mutations in the RPE65 gene. An adeno-assciated virus (AAV2) carries a functional copy of the RPE65 gene into the RPE cells, creating functional RPE65 protein, an enzyme that facilitates the conversion of all-trans-retinyl ester, the base fuel for phototransduction, to 11-cis-retinol, to restore the visual cycle. Patients treated with Luxturna have significantly better low luminance vision as demonstrated in the Multi-Luminance Mobility test1.
Personal Experience
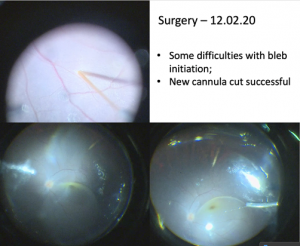
Our experience with Luxturna started just before the COVID-19 pandemic, with two sisters, aged 7 and 5 years, who displayed the characteristic features: a history of light staring, reasonably normal fundus appearances, albeit with white dots at the level of the RPE in the periphery and very limited fundus autofluorescence. There was reasonable retinal architecture on OCT, but the ellipsoid did have a slightly granular appearance.
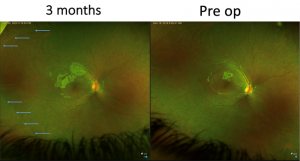
The older sister underwent gene therapy with Luxturna to the right eye. Her preoperative corrected visual acuity was 1.1 LogMAR in either eye. The improvement has been gradual, though she has commented on her better night-time vision. Her contrast sensitivity appears improved compared to her left eye, but with no preoperative measurements it is difficult to know the significance of this. Three months out, the acuity at both near and distance is one line better.
Her younger sister followed a very similar pattern, although she presented with better vision (0.68 LogMAR, N14). She had a greater degree of macular volume than her older sister. She had a larger than recommended volume of vector injected, but there are clear signs her vision is improving (0.56 LogMAR, N4.5 at three months). She reports that she doesn’t need the brightness turned up as much on her iPad in addition to her night vision getting better.
She also had what we presumed to be subretinal macrophages seen inferiorly. They melted away over the six-to-eight weeks following surgery. At three months there’s a subtle but nevertheless quite dramatic improvement with the white dots only seen in the far periphery. Her OCT retained its previous volume.
New studies are in progress utilizing antisense oligonucleotides or gene editing technologies such as CRISPR
Current Clinical Trials
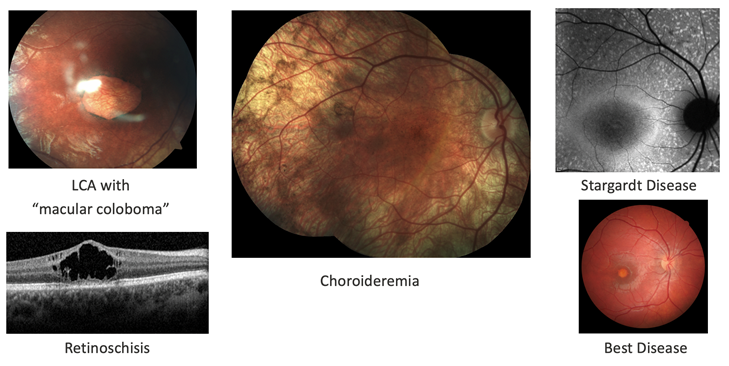
There are a lot of ongoing clinical trials for various IRDs including choroideremia, achromatopsia, X-linked retinitis pigmentosa, X-linked retinoschisis and CEP290 mutations, employing multiple treatment approaches ranging from adeno-assciated virus vectors to CRISPR in-vivo genome editing.
All of these studies need patients, so there is a need for patient registries, such as the My Retina Tracker from Foundation Fighting Blindness (https://www.fightingblindness.org/my-retina-tracker-registry) in the US, but other countries need to find ways to connect motivated patients to clinical trials and provide a means to access accurate genetic diagnostic reports.
These are exciting times for retinal genetics, but we must be cognizant that while we are at the dawn of a new era of treatments, we must remain realistic and not over-promise what may be deliverable in the future.
- Chung DC, McCague S, Yu ZF, et al. Novel mobility test to assess functional vision in patients with inherited retinal diseases. Clin Exp Ophthalmol. 2018 Apr;46(3):247-259. doi: 10.1111/ceo.13022. Epub 2017 Aug 31. PMID: 28697537; PMCID: PMC5764825.
LUXTURNA advisory board member and remuneration for lectures and conference presentations – Novartis; Key Opinion Leader – Alcon; Paediatric VR Instrumentation Advisory Board – DORC;
The Essential Role of Counselling

Hannah Scanga, MS, CGC
Lead Genetic Counselor
Dept. of Pediatric Ophthalmology, Strabismus, and Adult Motility
UPMC Children’s Hospital of Pittsburgh
Pittsburgh, PA, USA
hannah.scanga@chp.edu
Inherited retinal diseases (IRDs) in childhood vary significantly in age of onset and progression. In addition to clinical variability, the impact of IRDs can be variable on both the patient and the family and can contribute significantly to coping, adjusting, and accepting these diagnoses.
Psychological impacts can be quite vast and affect children and their parents very differently
Impacts of an IRD Diagnosis
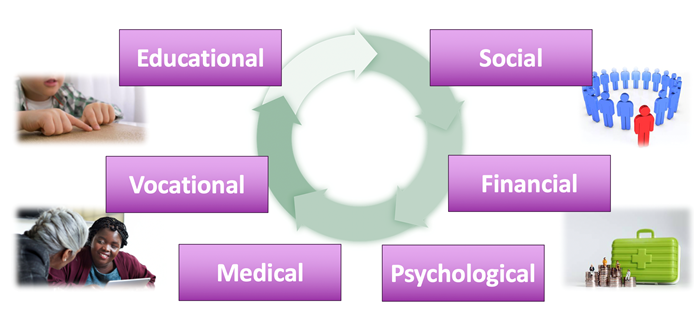
There are numerous impacts of receiving an IRD diagnosis. These impacts are interrelated and occur throughout the patient’s lifetime.
Educational – A child with an IRD diagnosis may perceive reduced access to education. To decrease this barrier, modifications are often needed, including increased font size or contrast, Braille instruction or assistive technology.
Social – While the child’s condition may not have an external sign medically, different aspects may show up in the classroom and in the child’s approach to learning, affecting peer relationships. Feeling like they don’t have the same access, interests or abilities can lead to social isolation or even discrimination and bullying between these children and their peers.
Financial – A diagnosis made in childhood results in later financial impacts. These may be related to medical costs and burden or to loss of future income based on opportunities or careers not pursued due to their vision.
Vocational – Also more impactful post-childhood, the vocational impact becomes an issue in choosing career pathways or access to different types of modifications that a workplace can provide.
Medical – Medical burden depends on the IRD and can peak at different times. Sometimes the medical burden is greatest after the initial diagnosis is made if there are a lot of exams, imaging, and testing. The burden can be more prominent in those with a syndromic retinal disease. Many different organ systems can be involved, and it may not be apparent at the time of diagnosis, meaning that the patient will go to multiple specialists for screening and evaluations.
The counselling that happens within different medical appointments informs patients of their prognosis, co-morbidities, management and treatment and research opportunities. Patients are more likely to comply and follow advice if they have a purpose, so counselling can help them understand the purpose of treatment, what they’re reaching for and what the goals are.
Psychological – Psychological impacts can be quite vast and affect children and their parents very differently. One of the first things that can happen in a child is a feeling of loss of independence. This is most prominent in teenagers. This cycles back into social isolation or discrimination. If the patient has a condition where their vision is impacted enough to use a cane or have some mobility modifications, or require the use of certain devices, it becomes a visible sign of their condition as a disability.
Parents of these patients often feel guilt and an effect on the familial relationships is very common. The parents may feel grief over their child’s loss; that their child is unable to pursue different opportunities, live a normal childhood, or be independent. They need to address the expectations they have for their children, which can be difficult. If the condition is progressive, these same feelings will re-emerge over time, as the condition gets worse and vision is further impaired.
Being aware of these impacts and thinking about someone who can help address the issues they create can lead to a very relevant referral to mental health specialists, therapists, or social workers.
Practice Points
There are several practice points for counselling patients with IRDs, regarding medical counselling or attention to patient burdens. One is having time to dedicate to the patient and the family, more than just providing a medical diagnosis with no supportive framework. Familiarity with the disease and the potential burdens can help address these issues and having relationships with extraocular specialists for medical needs, mental health needs or family-based counselling can be a big step.
Awareness of a patient’s ongoing and future educational and medical needs can also feel preventative and less reactive to patients, which may help them cope with their diagnosis. Being a resource or knowledge base for patients who are beginning to cope with, understand, and accept these diagnoses may lead to some additional adjustments. There are many patient advocacy groups that patients can connect with that act as an additional source of information that is frequently updated with new advances and research. Some advocacy groups also include avenues for patients and families to connect with each other, which can be helpful for these rare diseases.
Thinking of The Child as a Whole

Eduardo D. Silva, MD, PhD, FEBO
Pediatric Ophthalmology Department,
Centro Hospitalar Universitário Lisboa Central (CHULC), Lisbon
Centro Cirúrgico de Coimbra, Coimbra, Portugal
Hospital Lusíadas, Lisbon, Portugal
esilva2579@hotmail.com
Thinking of a child as a whole represents a major challenge for paediatric ophthalmologists. In the current era of molecular medicine, we want to make a correct diagnosis that will lead to appropriate medical management. Thus, it is important to establish translational and clinical collaborations that will allow us to reach a final diagnosis that will allow us to best help patients and their families.
The Basics
To reach an appropriate diagnosis, we need to ask questions and look at a child as a whole. Family history should always be considered to know if there are others in the family affected. Also think about interactions with genes and environment. It is not uncommon to identify severe nutritional deficiencies in children leading to vitamin A deficiency (no intake of green leafy vegetables, carrots and sweet potatoes), which causes a clinical phenotype that includes nyctalopia. This could be mistaken for a common retinitis pigmentosa.
It is possible now to explore genetic testing and segregation analysis. This is crucial because by having the correct genetic testing and very good clinical phenotyping we will be able to reach a final diagnosis. The diagnosis may change over time with the appearance of new signs and symptoms, and due to disease progression. Defining candidate selection for a certain treatment depends on where your patient is, thus the genetic identification in itself does not mean that you will be able to treat an individual.
Retinal Phenotypes in Metabolic Diseases
Retinal involvement may represent an important hallmark in metabolic diseases. Thus, for these genetic conditions, the ophthalmologist plays a central role in establishing the clinical diagnosis and monitoring disease progression and visual impact.
The image shown in Figure 10 represent the fundus of a child with LCHADD, part of the mitochondrial trifunctional protein, which metabolises long-chain fatty acids.
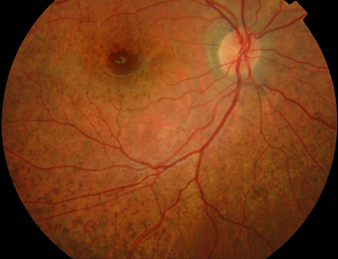
To reach an appropriate diagnosis in some cases, disease progression may lead to an evolving diagnosis
It is important to know this is an autosomal recessive condition caused by mutations in the HADHA gene and comprises a progressive chorioretinopathy that combines with bouts of rhabdomyolysis, cardiomyopathy and hepatomegaly. In this case, appropriate dietary restrictions may prevent disease progression and allow a very good quality of life.
Another example is Infantile Refsum disease, part of the Peroxisomal Biogenesis Disorders. It is a recessive condition caused by mutations in one of the PEX genes. Typical facial dysmorphism, developmental delay, hepatosplenomegaly, deafness, anosmia and a varying degree of pigmentary retinopathy may be present.
The hallmark of this condition is high levels of phytanic acid and very long-chain fatty acids. Again, appropriate dietary restrictions and management of metabolic stress may be crucial to prevent significant disease progression.
Evolving Condition Can Lead to Evolving Diagnosis
In some cases, disease progression may lead to an evolving diagnosis, with the appearance of new clinical/systemic findings, as in the case of a boy who presented first to our clinic at age 8 with progressive central loss of vision and changes in the central retina (bullseye maculopathy). He was diagnosed as having Stargardt disease, but one year later, when he started developing myoclonic seizures and showing increasing learning problems in school, we were able to reach the final diagnosis of juvenile neuronal ceroid lipofuscinosis (CNL3 gene), confirmed with conjunctival biopsy and appropriate genetic testing.
Reclassification
There are over 600 genes associated with eye disorders, some of which are related to retinal diseases. Such genetic knowledge has allowed better understanding of disease mechanism and reclassification of some retinal disease in the last few years. Some, which were initially thought only to cause a retinal condition, involve other organs and systems.
An example of a disease reclassification is ciliopathies. These conditions derive from mutations in genes essential for the normal structure and function of cilia; ciliopathies include Usher syndrome, affecting the eyes and hearing capacity, some forms of Leber Congenital Amaurosis, and Senior-Loken syndrome, where the retina and kidneys are affected, and Joubert syndrome, where eye and SNC involvement are identified. Appropriate diagnosis is important for design of treatments and establishment of a visual and life prognosis.